A review from the Committee for Orphan Medicinal Products (COMP) at the European Medicines Agency (EMA) has reported that “orphan designations” (OD) on inherited retinal degenerations (IRD) proposes a new approach to increase “the regulatory clarity, efficiency, and predictability for sponsors” on these regulatory applications. ODs provides a framework of incentives for medicines development on conditions or diseases with a prevalence that must not be more than 5 in 10,000 in the EU, slightly greater than the comparable process in the US. ODs were originally enacted in 2000, defined within Regulation (EC) No. 141/2000. An orphan designation, granted by the European Commission, gives marketing exclusivity in the EU for 10 years after approval. These incentives include trial protocol assistance, fee reductions, research grants, access to the centralised authorisation procedure and additional incentives for small and medium enterprises (SME).
In their current report, “IRDs” were traditionally driven by clinical / phenotypic features, as presented in the clinic however, as technological advances expand, the way by which IRDs were understood on the genetic aetiology and molecular biology, this presents challenges on how best to define therapeutic indications. As a result, the COMP have recently reviewed “the state of the art” in IRDs “to assess what would be the best approach or set of disease terms to use as the ‘condition’ for orphan designation in this therapeutic setting”. The review was based on the scientific literature and the experience of how EU orphan designations have been applied to date and this recruited IRD clinical experts, regulators and patients in order to build practical consensus.
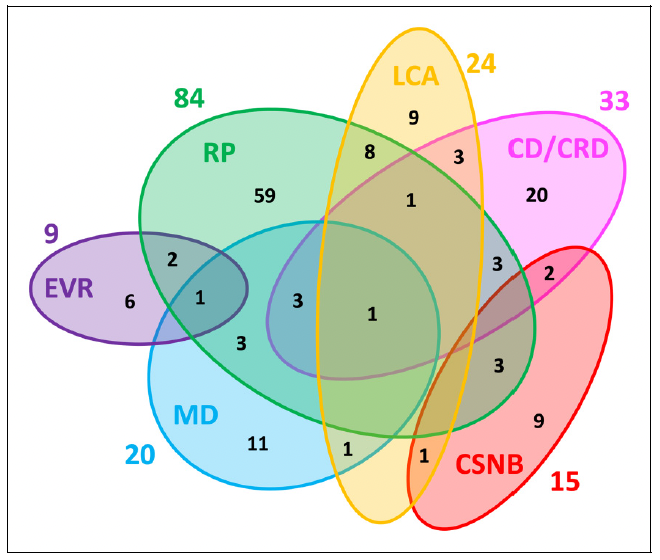
Figure 1. Genetic heterogeneity among the six major non-syndromic inherited retinal diseases (IRD). Numbers outside of the ellipses correspond to the number of non-syndromic IRD genes responsible for the specific disease, while numbers within the ellipses correspond either to disease-specific genes or to genes mutated in two or more diseases. The non-redundant total of genes associated with these non-syndromic IRD is 146.
RP: retinitis pigmentosa; LCA: Leber congenital amaurosis; CD/ CRD: cone dystrophy/cone-rod dystrophy; CSNB: congenital stationary night blindness; MD: macular dystrophy; EVR: exudative vitreoretinopathy. Reproduced unchanged with permission of authors Cremers FPM, Boon CJF, Bujakowska K, et al.7 © 2018 by the authors. Licensee MDPI, Basel, Switzerland under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Following deliberation by the clinical experts, regulators and patients on the literature review and expert consultation, COMP presented an optimised grouping for ODs with pharmacological approaches for in IRDs:

(Presented by Moseley et al, entitled “Inherited retinal dystrophies and orphan designations in the European Union”, Eur J Ophthalmol, 2024 Mar 18:11206721241236214. doi: 10.1177/11206721241236214. Distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 License (https://creativecommons.org/licenses/by-nc/4.0/) which permits non-commercial use, reproduction and distribution of the work without further permission provided the original work is attributed as specified on the SAGE and Open Access page (https://us.sagepub.com/en-us/nam/open-access-at-sage).
The COMP has supported that three options may be available for orphan designation concerning the condition as: (i) an amended set of OD groups for therapies that might be used in a broad spectrum of conditions; (ii) a gene-specific designation for targeted therapies, and (iii) an occasional term for products that do not fit in the above two categories. Commenting on the review, the researchers stated that, “overall, this three-pillar strategy is considered to provide a robust yet flexible framework for orphan designations in the complex field of the genetic spectrum underlying inherited retinal dystrophies. By discarding the more rigid andsomewhat out-dated phenotypically derived classification scheme traditionally used for IRD conditions, COMP now has the possibility of using a more agile decision matrix that allows for orphan designations adapted to the clinical state-of-the-art of the treatment under discussion. In conclusion, the new strategy of COMP regarding ODs in the domain of IRDs reflects regulatory adaptation to the evolution of our scientific understanding in the field of rare eye diseases and the development of innovative therapies. Therefore, if confirmed as appropriate with future regulatory experience, it is not excluded that a similar approach could be applied to other disease areas in the near future.