A consortium of researchers across Australia, New Zealand, USA, Italy, Portugal, Singapore, Netherlands, Taiwan and Canada, have reported a new study evaluating clinical, electrophysiological and genetic variants of inherited retinal diseases (IRD), all associated with mutations in the PRPH2 gene. PRPH2 gene were the fourth most common cause of IRD (5.2%) in the UK, however, there has been no clear genotype–phenotype correlations in disease, potentially “attributable to the relatively small sample sizes of previous clinical studies, variable penetrance, and no standardized system for phenotype grading using multimodal imaging”. The results of the current study, published in the journal IOVS (Invest Ophthalmol Vis Sci., 2024;65(5):22), have concluded that six distinct fundus auto-fluorescene (FAF) phenotypes are now associated with PRPH2 variants, and one FAF phenotype may have multiple ERG phenotypes, “demonstrating a discordance between structure and function. Aside from the important novel biological findings, the researchers have proposed that the information may be useful for future clinical trials.
According to the researchers, there are 352 disease-causing variants in PRPH2 listed in the Human Gene Mutation Database with autosomal dominant, recessive and digenic inheritance. PRPH2 encodes peripherin, a photoreceptor specific tetraspanin membrane protein, required for the formation and maintenance of rod and cone outer segments (OS). Dominant variants in PRPH2 lead to central areolar choroidal dystrophy (CACD), retinitis pigmentosa, pseudo-Stargardt pattern dystrophy, butterfly pattern dystrophy (BPD) and vitelliform macular dystrophy (VMD). In addition, the literature has been reported in pedigrees with marked interfamilial and intrafamilial variability and penetrance. In the international consortium’s study, 241 patients were recruited from 168 families across 15 sites in 9 countries, each participant with pathogenic or likely pathogenic variants in PRPH2.
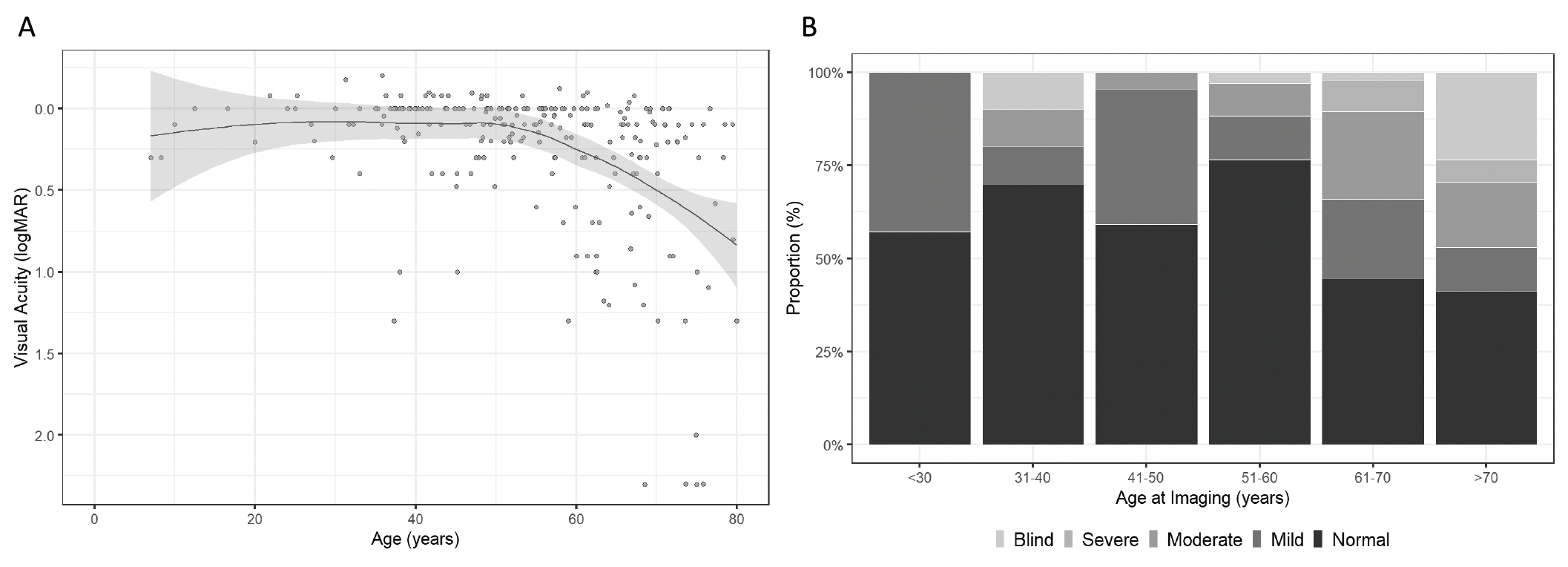
Fig 1. (A) Locally estimated scatterplot smoothing (LOESS) curve using VA from the better-seeing eye illustrating the average evolution of VA with age across the entire cohort. (B) Proportion (%) of PRPH2 patients with normal, mild (VA, <20/40 and ≥20/60), moderate (VA, <20/60 and ≥20/200), severe visual impairment (VA, <20/200 and ≥20/400) and blindness (VA, <20/400) by age at imaging.. (This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License; Heath Jeffery RC, Thompson JA, Lo J, et al. Retinal dystrophies associated with peripherin-2: genetic spectrum and novel clinical observations in 241 patients. Invest Ophthalmol Vis Sci. 2024;65(5):22. https://doi.org/10.1167/iovs.65.5.22)
The report of the work showed 22 novel pathogenic variants in one of the largest multi-centre case series and their results recorded the median age at symptom onset of 40 years (range, 4–78 years). FAF phenotypes included normal (5%), butterfly pattern dystrophy, or vitelliform macular dystrophy (11%), central areolar choroidal dystrophy (28%), pseudo-Stargardt pattern dystrophy (41%), and retinitis pigmentosa (25%). The median visual acuity was 0.18 logMAR (20/30 or 6/9) (interquartile range, 0–0.54 logMAR) and 0.18 logMAR (interquartile range 0–0.42 logMAR) in the right and left eyes, respectively. ERG showed a significantly reduced amplitude across all components (P < 0.001) and a peak time delay in the light-adapted 30-Hz flicker and single-flash b-wave (P < 0.001). Of 91 different PRPH2 variants, there were 21 frameshift, 12 nonsense, 2 exon deletions, 4 splice-site, and 1 start–loss, of which most were expected to result in loss of function or haploinsufficiency. Reporting on the results, the researchers commented that, “although the severity of PRPH2-associated IRD can vary considerably, VA often remained relatively stable until the fifth decade, with a constant decline thereafter. There was a range of retinal presentations including five distinct FAF phenotypes. Within each FAF phenotype group, there was often a spectrum of electrophysiological changes. The detailed clinical and genetic information provided here will be useful for clinicians to aid their work-up of patients with a PRPH2-associated IRD”.