Researchers at the Department of Optometry and Vision Sciences, and the Ophthalmology Department of Surgery, based in the University of Melbourne, Australia, has reported results of a cross-sectional study of RPGR carriers among different retinal phenotypes of X-linked retinitis pigmentosa (XLRP). In their study, published in the journal Genes (2025, vol. 16(2), 221), they showed that female RPGR carriers with severe retinal phenotypes “had significantly decreased visual function and changes in retinal structure, in comparison to both the controls and carriers with mild retinal disease.” These phenotypes may be useful as outcome measures for RPGR carriers which have a range of variations of disease. According to the researchers, they noted that “eight gene therapy clinical trials for RPGR associated XLRP registered on clinicaltrials.gov; however, females are only eligible to enrol in two of these studies.”
XLRP affects approximately 1 in 15,000 people worldwide, which primarily affects males, but female carriers may also experience a range of mild-to-severe progressive visual impairments. The disease is caused by pathogenic variants in at least three genes: retinitis pigmentosa guanosine triphosphatase regulator (RPGR), activator of Arl3 GTPase (RP2), and orofaciodigital syndrome I (OFD1). Over 300 mutations occur in the RPGR gene, most of which in the open reading frame exon (ORF15), causing an abnormally short protein that is expressed in the connecting cilium of photoreceptors, and is an important component of all ciliated cells in the body. Up to 73% of mutations occur in the ORF15 region, which is regarded as the mutational hotspot of the RPGR gene due to a glutamate–glycine-rich domain at this site, leading to genomic instability.
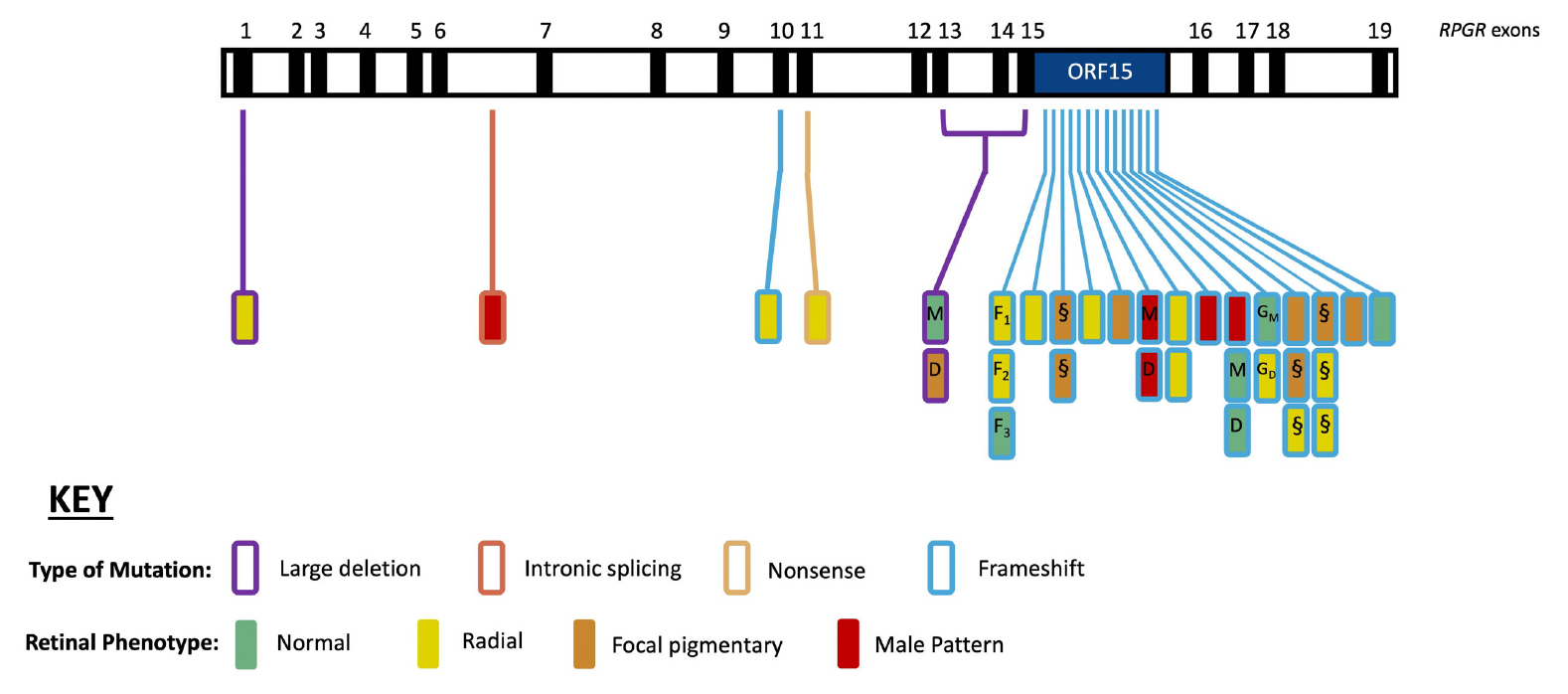
Figure 1. Retinal disease phenotype and genetic variants along the RPGREx1–19 gene. Exons 1–19 of the RPGR gene are depicted, with each rectangle denoting a female RPGR carrier. Vertically stacked rectangles illustrate carriers with the same disease-causing variant, and family members are depicted as follows: D, daughter; GM, grandmother; GD, granddaughter; M, mother; S, sister. Note: F1, F2, and F3 are members of the same extended family (i.e., F1 is the aunt, and F2 and F3 are first cousins but not daughters of F1). The location and colour of the lines indicate the affected exons and type of mutation, respectively. The colour of the rectangle (filled) indicates the retinal disease phenotype (normal, radial, focal pigmentary, and male pattern phenotypes). This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/), entitled: Retinal Disease Variability in Female Carriers of RPGR Variants Associated with Retinitis Pigmentosa: Clinical and Genetic Parameters, by Gocuk, SA et al.; Genes, Volume 16, Issue 2, 10.3390/genes16020221.
Mutations in the RPGR gene can be associated with a rod-cone or cone-rod dystrophy phenotype. The most common presentation is as a rod-cone dystrophy. It is one of the most severe forms of RP with nyctalopia and progression to legal blindness by the third to fourth decade. The disorder is initially identified with difficulties in scotopic visual function, where there is a predominant loss of rod photoreceptors.
Retinal disease has been classified into four retinal disease severities based on FAF imaging, as defined by a previous study in 2018 (Nanda et al. Genes 2018, 9, 643): normal (N), radial (R) pattern reflex without pigmentary retinopathy, focal (F) pigmentary retinopathy, and male (M) phenotype.
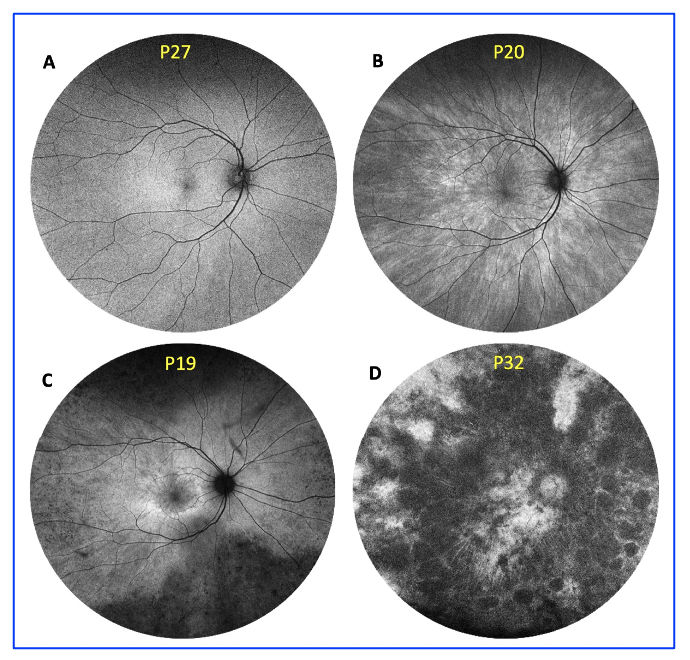
Figure 2. Retinal disease severity of RPGR carriers. Retinal disease classification was based on fundus autofluorescence imaging using Optos ultrawide retinal photography, as previously described by Nanda et al. (2018): (A) normal; (B) radial; (C) focal pigmentary retinopathy; and (D)male phenotype. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/), entitled: Retinal Disease Variability in Female Carriers of RPGR Variants Associated with Retinitis Pigmentosa: Clinical and Genetic Parameters, by Gocuk, SA et al.; Genes, Volume 16, Issue 2, 10.3390/genes16020221.
In their recent study, thirty-five female RPGR carriers and thirty healthy controls were recruited, collating measurements on refractive error, BCVA, LLVA (low luminance visual acuity) and LLD (low luminance deficit). On average, RPGR carriers with focal pigmentary retinopathy had a greater myopic refractive error compared to the healthy controls (approximately 3 dioptres more myopia, p < 0.01). BCVA was significantly reduced in RPGR carriers with focal pigmentary retinopathy or male pattern phenotypes compared to those with milder phenotypes (p < 0.05) and healthy controls (p < 0.001). In summary, “a comprehensive evaluation of genotype–phenotype correlations revealed that although there may not be a direct link between the genotype and the severity of the retinal disease phenotype, RPGR carriers with disease-causing variants at the ORF15 site appeared to have poorer clinical outcomes, particularly concerning LLVA and retinal thickness, compared to carriers with exon 1–14 variants, irrespective of age.” These results may be considered as relevant outcome measures for identifying eligible female patients for participation in future clinical trials and/or the efficacy of treatments.