Clinical researchers at the Department of Ophthalmology and Johnson Retina Centre, University of Washington, Seattle, have reported on the clinical characteristics, natural history, genetic landscape, and the phenotypic spectrum of neuronal ceroid lipofuscinosis (NCL)-associated retinal disease. The research, also supported by Seattle Children’s Hospital and Children’s Hospital Los Angeles, have shown that underlying genetic variants appear to drive phenotypic diversity in different forms of NCL. Gene testing may provide molecular diagnosis and expand the management and support for children and their families. In their clinical study, the researchers have concluded that, “with intravitreal enzyme replacement therapy on the horizon as a potential treatment option for NCL-associated retinal degeneration, precise structural and functional measures will be required to more accurately monitor disease progression.”
Neuronal ceroid lipofuscinoses (NCL) comprise a group of genetically heterogeneous lysosomal storage disorders are the most common inherited neurodegenerative disorders of childhood, primarily arising from an autosomal recessive pattern of inheritance. Thirteen (13) causative genes are highly relevant for NCL characterized by the accumulation of auto-fluorescent storage material in neurons, leading to epileptic seizures, progressive psychomotor retardation, and premature death. Abnormal accumulation of pathology leads to generalized photoreceptor disease and profound visual impairment. In the current study, researchers evaluated 12 subjects with pathogenic, or likely pathogenic variants, in 5 NCL-causing genes (CLN3, TPP1, PPT1, CLN6, and MFSD8). Patients presented with varying phenotypes ranging from severe neurocognitive features (n = 8; 67%), including seizures and developmental delays and regressions, to non-syndromic retinal dystrophies (n = 2; 17%). Visual acuities at presentation ranged from light perception to 20/20. In those with recordable ERGs, the traces were electronegative and suggestive of early cone dysfunction. Fundus imaging and OCTs demonstrated outer retinal loss that varied with underlying genotype.
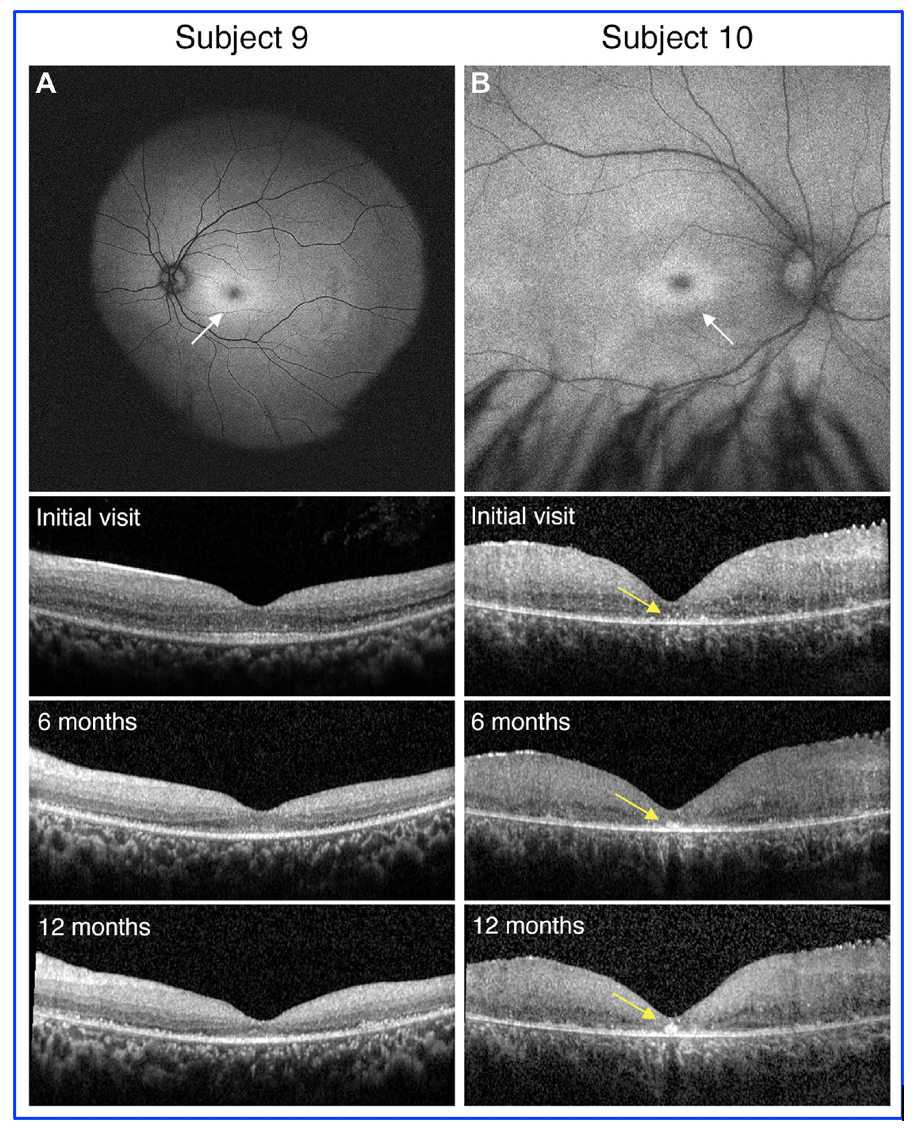
Figure 1. Genotypic variants of PPT1 can lead to differential retinal phenotypes and progressive features. A, In Subject 9 with classic systemic features of neuronal ceroid lipofuscinoses, the exam was consistent with a bulls eye maculopathy as evidenced on fundus autofluorescence (white arrow). Over the course of 1 year follow-up, OCT findings showed progressive loss of the outer retinal layers which correlated with decline in best-corrected visual acuity. B, In subject 10 with more isolated retinal findings and genotypic findings consistent with granular osmiophilic deposit, the exam was again consistent with a bulls eye maculopathy as evidenced on fundus autofluorescence (white arrow), but OCT findings were most notable for almost total loss of the outer retinal bands with hyperreflective foci in the fovea that progressively enlarged over the course of 12 months (yellow arrows). [This work is an open access content, licensed under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/), authored by Huey, J et al., entitled, “Genetic Reasons for Phenotypic Diversity in Neuronal Ceroid Lipofuscinoses and High-Resolution Imaging as a Marker of Retinal Disease”, published in Ophthalmology Science, Vol. 4, No. 6, Dec 2024, https://doi.org/10.1016/j.xops.2024.100560].
Following their analysis from the cohort, the study “provides new insight into how individual genotypes can influence phenotypic features of retinal disease in different forms of NCL.” The researchers found that subjects had molecular confirmation of either homozygous or compound heterozygous pathogenic, or likely pathogenic variants in PPT1, TPP1, and CLN3. In addition, 5 novel pathogenic or likely pathogenic variants were identified in their cohort, including TPP1 c.837C>G, TPP1 c.508þ4A>G, CLN6 c.247dup, MFSD8 c.1217_1218dup, and MFSD8 exon 11e13 deletion. The clinicians also commented that, “understanding the genetic contributions to disease can have important retinal implications that can help guide the clinician. In certain cases, it may differentiate between a classic, multisystemic NCL presentation and a more isolated retinal phenotype.” Pre-clinical trials for specific therapies for TTP1-related NCL are in progress in animal models, reportedly preserving retinal function until reached end-stage neurological disease. Further research are likely to progress on a range of technological strategies to support these NCL patients and their families.