Researchers at the Eye Centre, Zhejiang University School of Medicine, Hangzhou, China, have reported a novel truncating mutation in the RP1 gene (retinitis pigmentosa 1), not identified previously in the literature. The researchers showed that a heterozygous frameshift in the RP1 gene may trigger the autosomal dominant retinitis pigmentosa (adRP) pathology, causing the loss of functional protein domains. Their findings broaden the genetic mutation spectrum of RP1 in adRP cases, and the relationship between genotype and phenotype will provide further support for diagnostic and research treatments for patient care.
More than 90 genes, including thousands of mutations, have been linked to retinitis pigmentosa (RP), one of which, RP1, encodes the photoreceptor-specific microtubule-associated protein residing in the outer segment of rods and cones. RP1 functions for the stability and organization of the outer segment discs and RP1-autosomal dominant RP (adRP) manifests nyctalopia and decreased peripheral vision in their 20s and 30s with near-normal visual acuity (VA) into their 50s and 60s. In contrast, autosomal recessive RP (arRP) inheritance pattern typically manifests before 10 years of age, and is characterized by secondary macular involvement that can result in legal blindness by age 20 years. RP1 is estimated to cause up to ~11% of each adRP and arRP.
In their recent research in Zhejiang University School of Medicine, 27 patients were enrolled from a family spanning 5 generations. The study found the previously undocumented truncating mutation c.2015_2018del p. (Lys672Argfs*9), within the RP1 gene, had concomitant subluxated lens and posterior subcapsular cataract.
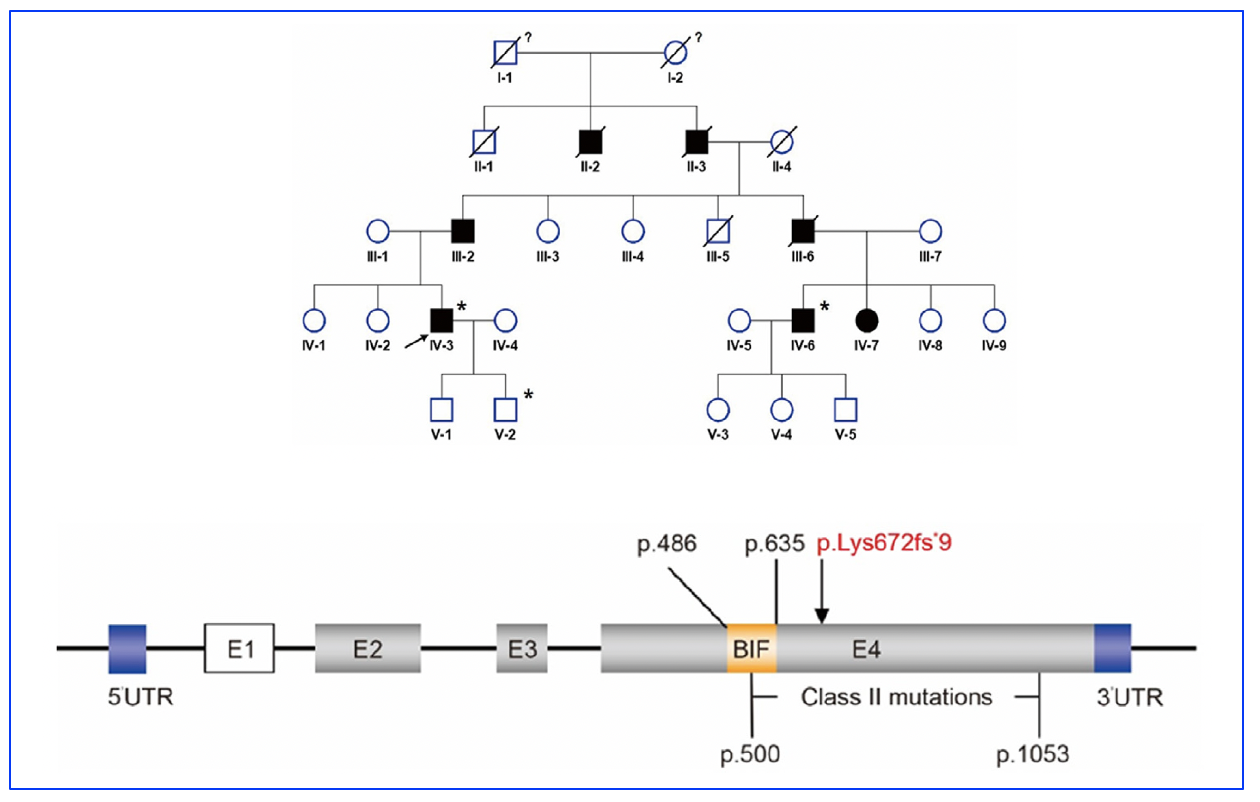
Figure 1: Pedigree of the RP patient’s family. Segregation analysis of RP1 mutation in a pedigree represents an autosomal dominant inheritance pattern. Squares and circles represent males and females respectively. Closed and open symbols indicate affected members and unaffected subjects. The arrow denotes the proband. Family history was negative for consanguinity. The slash symbol indicates that the subject was deceased. The asterisk shows the individual underwent both clinical and genetic analyses. The question mark indicates the ophthalmic history is not available.
RP1 gene and identified mutations. The novel mutation of RP1 gene c.2015_2018del p. (Lys672Argfs*9) identified in this study belongs to Class II mutations (p.486-p.635). E -exon, UTR – untranslated region, BIF – the Drosophila melanogaster domain.
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/), entitled: A novel RP1 truncating mutation that causes autosomal dominant retinitis pigmentosa (ADRP), by W. Wu et al. published in Advances in Ophthalmology Practice and Research, 5 (2025) 41–48.
Their report commented that, “analysis indicated that the novel mutation affects highly conserved sites that likely play crucial roles in essential physiological functions. Furthermore, analysis of the secondary structure revealed that the truncating mutation results in the loss of most functional protein domains in a clear and evident manner”. The RP1 variant is likely to perturb the structure and orientation of the cilia in rods and cones however, it is yet to be fully detailed on how the specific molecular pathology functions and how the truncated proteins may interfere signal transmission within photoreceptors. The researchers stated that, “these truncated proteins have the potential to impair photoreceptor function, leading to cell death and exacerbating the progression of RP”. It will be interested to learn if pharmacological or genetic strategy may be blocked to ameliorate the pathology and further research will likely await over the coming years.