Researchers at the Biosciences Institute, Newcastle University, UK, have reported results from a study on differentiated retinal organoids derived from Stargardt disease patients (STGD1). The study evaluated key retinal neurons and photoreceptors with outer segments positive for ABCA4 expression and their results “highlight the suitability of retinal organoids in STGD1 modelling. Their ability to display genotype-phenotype correlations enhances their utility as a platform for therapeutic development”. As there are an estimated >150,000 individuals worldwide with Stargardt disease, and with no treatments available, there is a significant demand to further define genotype and phenotype details for inherited retinal degeneration, not least of all proposing new outcomes for clinal research endpoints.
STGD1 is an inherited disorder that can cause vision loss in children and young adults, affecting approximately 1:8000 individuals worldwide. The disorder is caused by variants in the photoreceptor-specific ABCA4 gene, which encodes the adenosine triphosphate-binding cassette, subfamily A, member 4. The gene encodes a transporter protein within retinal photoreceptor cells facilitating the active transport of potentially toxic retinoid compounds removing toxic by-products from the visual cycle. A number of pathogenic variants within certain introns of the ABCA4 gene may result aberrant splicing of the gene – including pseudogenes – essentially non-functional segments of DNA. Loss of the protein results in the accelerated dimerization of vitamin A, forming toxic by-products that irreversibly damage the retina, resulting in progressive vision loss. According to the UK researchers, “ABCA4 contains 50 exons in a 128-kb region gene which boasts an expansive and ever-growing list of genetic variation. Over 1,000 pathogenic/ likely-pathogenic variants have thus far been reported in both coding and non-coding regions of the gene.”
Retinal organoids (ROs) act as a miniaturized and “simplified” version of a retina, providing key functional, structural, and biological activities capable to self-organise in three dimensional culture. The organoid is used to study development and disease in the lab, including research pathology and drug discovery, gene and cell therapies, tissue engineering and regenerative medicine. In their recent study at Newcastle University, researchers generated induced pluripotent stem cells (iPSCs) from two monoallelic late-onset STGD1 cases and differentiated them into retinal organoids (ROs) alongside affected and unaffected control iPSCs. They showed that STGD1 ROs exhibited a phenotypic defect characterised by photoreceptor mis-localisation, which correlated in intensity with disease severity. In addition, single-cell RNA sequencing data showed that STGD1 ROs displayed gene dysregulation in stress-response and phototransduction pathways, “indicating that the model can recapitulate STGD1 phenotypes in vitro. Most importantly, we identified the missing alleles in two STGD1 patients using whole genome sequencing (WGS) and long-read RNA sequencing (LRS) technologies.”
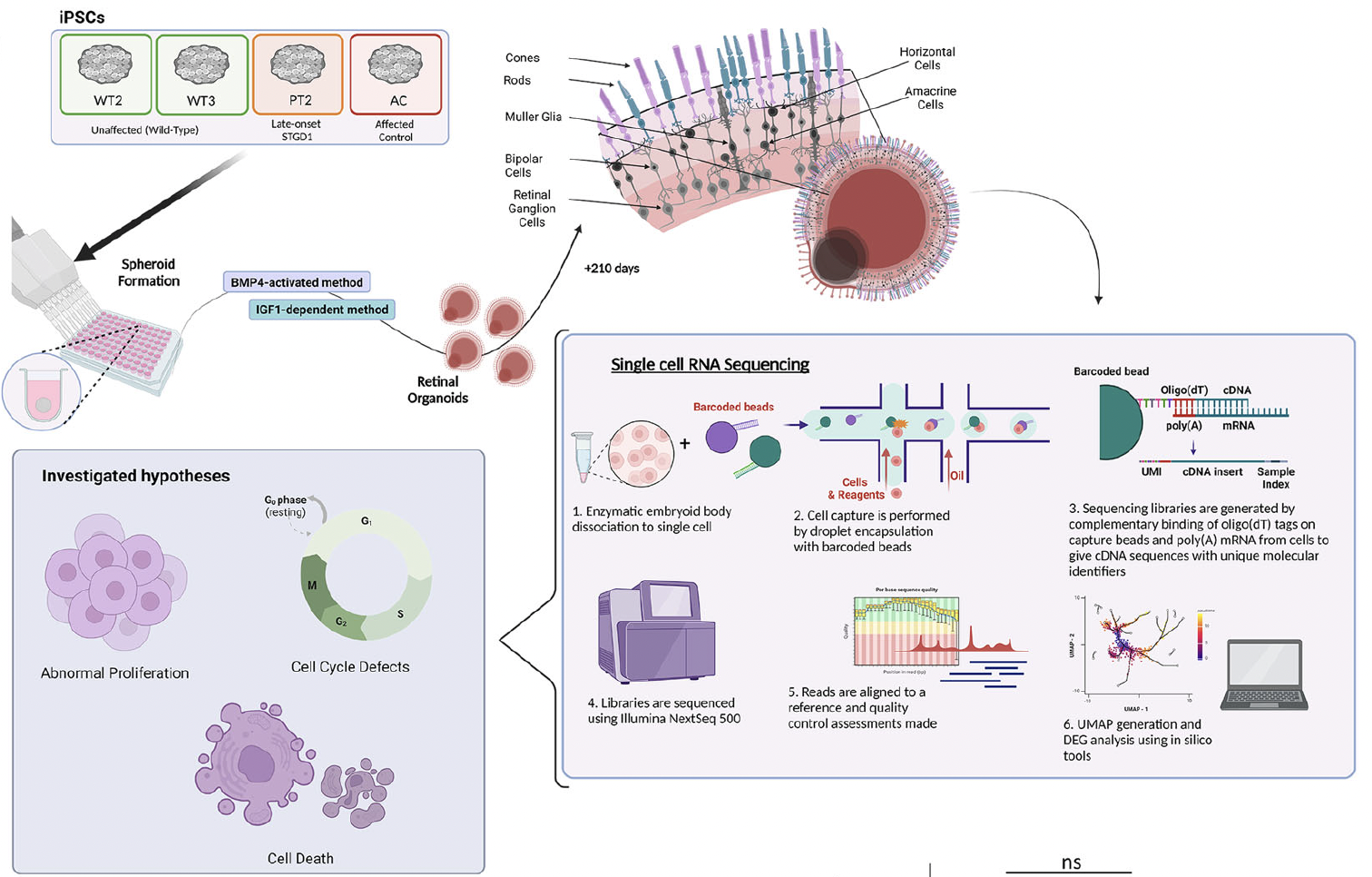
Figure. 1 scRNA-Seq shows pathomechanism of photoreceptor mislocalisation to be likely secondary to activation of stress-response in STGD1 ROs. A Schematic depicting the scRNA-Seq experimental process. Briefly, ROs are derived from iPSCs and matured to Day 200 where all retinal neurons are expected to be developed. scRNA-Seq library prep is conducted on RNA transcripts from dissociated single cells of the STGD1 ROs and barcoded mRNA transcripts are sequenced using Illumina NextSeq 500 platform. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/), entitled: Unravelling genotype-phenotype correlations in Stargardt disease using patient-derived retinal organoids, by Watson, A et al.; Cell Death and Disease (2025) 16:108 ; https://doi.org/10.1038/s41419-025-07420-7.
In summarising the results of their study, the researchers commented that, the data “has achieved genetic resolution for previously monoallelic cases of STGD1, enabling these individuals to participate in clinical trials for emerging treatments. Importantly, we demonstrate that genotype phenotype correlations are possible in vitro, with variant-induced molecular pathology, making this model vital for drug development, in vitro toxicology, and further disease modelling.”