Researchers at the Ophthalmic Genetics and Visual Function Branch, National Eye Institute, Bethesda, Maryland, USA have reported a genetic association between UBAP1L variants and retinal dystrophy. Clinical and genetic evidence showing the loss of UBAP1L function, expressed from a 1,707 bp of mRNA, was associated with inherited retinopathy in humans. According to the researchers, none of the patients from the study had a family history of IRDs, and a subsequent detailed research study found four (4) homozygous UBAP1L variants identified in six (6) individuals from six (6) unrelated families affected by variable forms of retinal dystrophy. Putative loss-of-function variants were found to be rare within the general population and the loss of UBAP1L function is associated with non-syndromic retinal dystrophy, highlighting its previously unrecognized roles in human health.
The six-patients with IRDs came from four tertiary hospitals – National Eye Institute, National Institutes of Health Clinical Center USA, Moorfields Eye Hospital, Royal Liverpool University Hospital and NHS Birmingham Women’s & Children’s hospital, UK. Four homozygous UBAP1L variants were identified in the affected individuals from six families (with 2 frameshift variants, 1 canonical splice variant and 1 noncanonical splice variant). Participants presented with retinal dystrophy including maculopathy, cone dystrophy, and cone-rod dystrophy. The protein – ubiquitin-associated protein 1 like (UBAP1L) – is found on chromosome 15q22.31. The UBAP1L encodes one known protein-coding transcript consisting of six exons that encodes the 381 amino acids. The exact function of UBAP1L is unknown however, it is predicted to enable ubiquitin binding activity and to be a part of Endosomal Sorting Complex Required for Transport-I complex (ESCRT-I). The UBAP1L expression appears to be highly enriched in the retina and retinal pigment epithelium (RPE) and a comparison between human and mouse orthologs show approximately 70% identity.
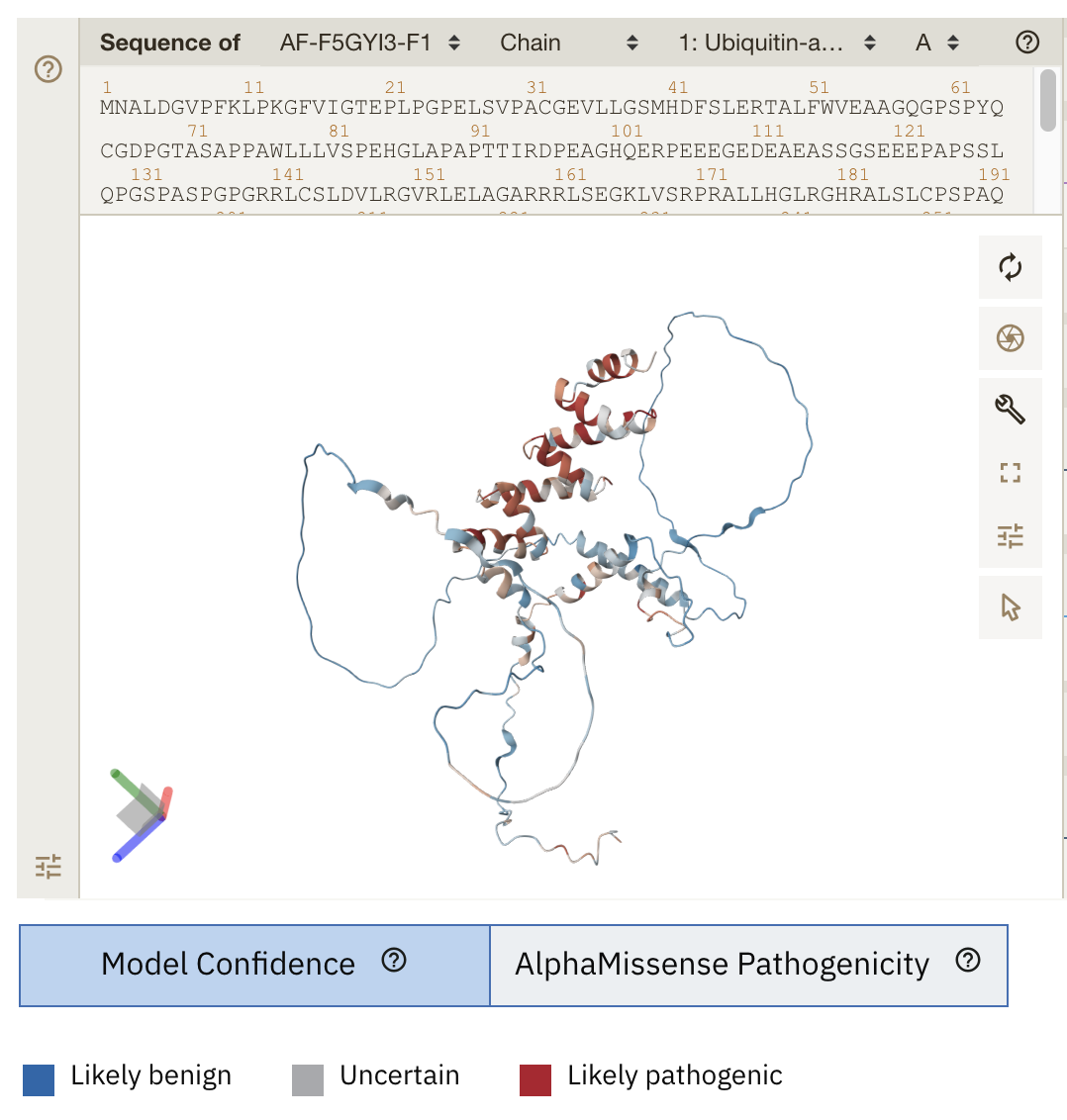
Figure 1. The novel work displayed at EMBL’s European Bioinformatics Institute (https://alphafold.ebi.ac.uk/entry/F5GYI3) which shows a 3-D model for UBAP1L with each colour for each residue as the average AlphaMissense pathogenicity score across all possible amino acid substitutions at that position. Likely pathogenic residues are in red. [Data is available for academic and commercial use, under a CC-BY-4.0 licence].
Human UBAP1L showed high expression levels in cone and RPE cells, while in contrast, the mouse Ubap1l showed high expression in cones but at a lower level in RPE cells. The UBAP1L expression was observed at low levels in the rods of both human and mouse retinas. Researchers then generated a knockout mouse model of the gene by removing the last 3 exons, which was expected to disrupt the function however, the research found no evidence of retinal degeneration in the knockout animals up to 15 months of age.
A co-senior author on the paper, Laryssa A. Huryn, M.D., an ophthalmologist at the NEI, part of the National Institutes of Health, commented that, “these findings highlight the importance of providing genetic testing to our patients with retinal dystrophy, and the value of the clinic and lab working together to better understand retinal diseases”. In conclusion of the valuable research results, the data from their publication provides “strong clinical and genetic evidence supporting that loss-of-function variants in UBAP1L were associated with autosomal recessive retinal dystrophy (macular, cone, and cone-rod dystrophies) presenting in early adulthood. The retinal degeneration is progressive, resulting in a severe loss of photoreceptors and visual field in late adulthood”. The detailed study is published in the JAMA Ophthalmol, on Sept. 26th, 2024.