Researchers at Janssen Pharmaceuticals N.V. and the ERN-EYE Network at the Center for Retinitis Pigmentosa of Veneto Region, Camposampiero Hospital, Padua, Italy, have reported results from a cross-sectional physician survey on X-kinked retinitis pigmentosa (XLRP), including patient pathways in European countries. Researchers aimed to evaluate the real-world burden of XLRP from the perspective of retinal specialists and geneticists in five countries – France, Germany, Italy, Spain, and the UK. According to the study, the diagnosis, genetic testing, and management pathways among XLRP patients may vary considerably. Respondents reported that “their patients had long and complex diagnostic journeys that varied among countries, including multiple potential referral pathways, up to 10 years between onset of symptoms and diagnosis, and a wait of between 6 weeks and 6 months for diagnosis confirmation by genetic testing”. As a result, the authors commented that, “there is a need for more standardized diagnosis and management pathways, and QoL [quality of life] assessments, due to the major impact that XLRP has on patients’ lives”.
XLRP is an incurable genetic disease that causes blindness in males affecting approximately one in 15,000 people. The disease is caused by pathogenic variants in two genes, retinitis pigmentosa guanosine triphosphatase regulator (RPGR) and RP2, accounting for approximately 70% and 20% of cases. Over 300 mutations occur in the RPGR gene, most of which in the open reading frame exon (ORF15), causing an abnormally short protein that is expressed in the connecting cilium of photoreceptors, and is an important component of all ciliated cells in the body. Mutations in the RPGR gene can be associated with a rod-cone or cone-rod dystrophy phenotype. The most common presentation is as a rod-cone dystrophy. It is one of the most severe forms of RP with nyctalopia in most affected males before 10 years of age and progression to legal blindness by the third to fourth decade. The disorder is initially identified with difficulties in scotopic visual function, where there is a predominant loss of rod photoreceptors. Simultaneously, peripheral vision deteriorates, resulting in visual field constriction on perimetry findings. The majority of cases present with a rod-cone dystrophy-type disease progression, where central visual acuity is initially less impaired than the peripheral field loss. The fovea is ultimately affected in all cases during the late stages of the disease by subsequent cone photoreceptor degeneration.
In their current cross sectional study, entitled “EXPLORE”, respondents reported that patient independence decreased over time, where 37% of patients were considered “completely autonomous” at diagnosis, versus 23% at the last consultation. According to their data, at their last visit, 45% of patients were active in the workforce; 67% (12/18) of “completely autonomous” patients had active working status compared with 13% (1/8) of “completely dependent” patients. The average time from onset of symptoms to diagnosis was 4 years and varied among countries. In 78% of patients, XLRP was confirmed by genetic testing, the rate of which varied among countries (range, 50-94%), taking up to 6 months to receive results. Specialists identified unmet needs in XLRP management including more standardized assessments of quality of life (QoL) as well as easier and earlier access to specialists, genetic testing, patient support programs, and effective treatment options. The report indicated that, “in Germany and the UK, patients were reported to have seen retina specialists/geneticists through multiple steps and routes, including referrals by general practitioners, optometrists, and generalist ophthalmologists. In France, Italy, and Spain, patient pathways were reported to be more linear; most patients had seen retina specialists/geneticists as a result of referral by generalist ophthalmologists. Retina specialists reported seeing patients with XLRP typically once or twice a year for consultation”.
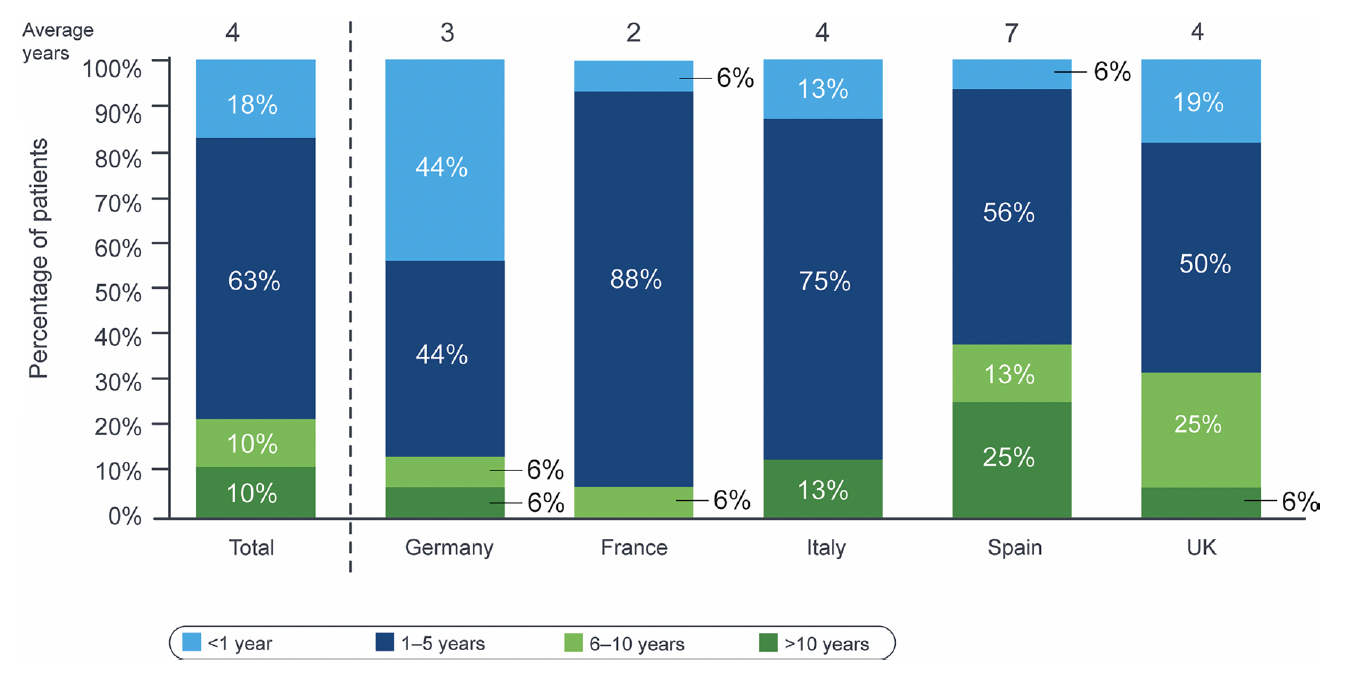
Figure 1: Time to diagnosis: average duration in years between the onset of symptoms and diagnosis. Total, N = 80; country, N = 16.
Open Access article is licensed under a Creative Commons Attribution 4.0 International License; Pungor et al., Impacts of X‑linked Retinitis Pigmentosa and Patient Pathways in European Countries: Results from the Cross‑sectional EXPLORE XLRP‑1 Physician Survey, Advances in Therapy (Springer), Volume 41, pages 3378–3395, (2024).
In summary, the researchers concluded that, “those unmet needs include faster diagnosis, more standardized QoL assessment, improved and earlier access to patient support/rehabilitation programs, and effective treatment options. Additional needs include policies that provide better access to employment, streamlined patient pathways that enable earlier diagnosis and management, and broader access to genetic testing with faster delivery of testing results. Addressing these unmet needs and standardizing the diagnosis and management pathways across countries should be a priority for healthcare systems to ensure early diagnosis and effective management of patients with XLRP”.