Clinical researchers at the Sorbonne Université, INSERM, Institut de la Vision, Paris, have reported multiple pathogenic variants in the RP1L1 gene, associated with autosomal dominant, autosomal recessive, autosomal recessive rod-cone dystrophies and occasionally, pseudovitelliform maculopathy. The study was designed to explore the natural history of RP1L1-related retinopathy, and uncover novel genotype – phenotype correlations. Prof. Isabelle Audo, the lead author of the study, stated that, “our approach allowed for a careful reappraisal of patients presenting with atypical phenotypes and highlighted the complex spectrum of RP1L1-associated retinopathy”. The results have provided a comprehensive valuable analyses and resource to assist other clinicians that may wish to evaluate such patients from RP1L1 variants.
Retinitis pigmentosa-1-like-1 (RP1L1) gene encodes a component of the photoreceptor axoneme in the morphogenesis and maintenance of photoreceptor outer segments (OS) and the gene spans ~105 kilobases on chromosome 8p23.1. The protein product comprises 2,400 amino acids, expressed in rod and cone photoreceptors. The most common phenotype linked to pathogenic variants in this gene is occult macular dystrophy (OMD), an autosomal dominant macular dystrophy with normal fundus appearance and reduced macular function, documented by multifocal electroretinogram (mfERG). Other variants resulting in OMD are found in a hotspot region between amino acid residues 1196 and 1201, a region of unknown protein domain, highlighting the significant gaps in the understanding of RP1L1. In addition, variants of RP1L1 can cause recessive rod-cone dystrophy, typically resulting from the association of two null variants. The distribution of RP1L1 variants identified, including those associated with RP and atypical presentations were reported in the literature, summarized below.
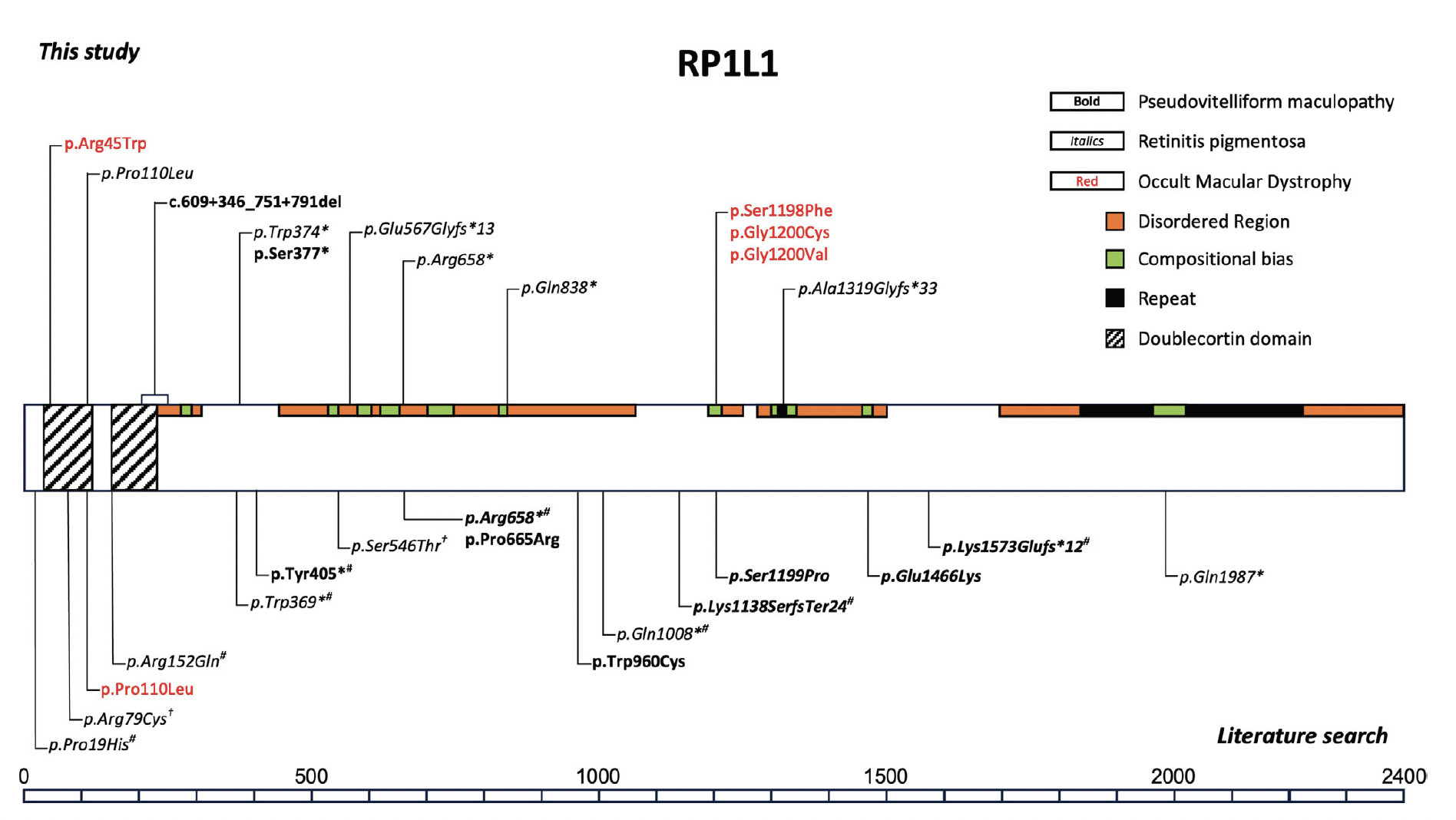
Figure 1. Schematic representation of the variants in the RP1L1 gene observed in our cohort (upper section). Variants associated with typical and “atypical” occult macular dystrophy (OMD) are in red, retinitis pigmentosa (RP) are in italics, and pseudovitelliform maculopathy is in bold. A literature review (PubMed: accessed August 30, 2024) was conducted to identify all published variants in the RP1L1 gene associated with autosomal recessive RP or pseudovitelliform maculopathies and related phenotypes (lower section). With the exception of p.(Pro110Leu), additional variants associated with OMD in literature are not included in this figure. †Homozygous or compound heterozygous with a missense variant. #Homozygous or compound heterozygous with a null variant.
[This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, authored by Antropoli et al., entitled, “Phenotypic and Genotypic Characterization of RP1L1-Associated Retinopathy”, published in Investigative Ophthalmology & Visual Science, April 2025, Vol. 66, 7. doi:https://doi.org/10.1167/iovs.66.4.7].
The outcomes from the retrospective study were assessed by 20 patients in France, from European descent (45%) and African descent (25%), with the remaining (30%) had an unknow ethnic descent. The study included 20 patients (40 eyes) from 19 families: 12 (60%) with OMD, 4 (20%) with RP, and 4 (20%) atypical cases (3 “non-occult” macular dystrophy, 1 rod-cone dystrophy with vitelliform maculopathy). In their cohort, autosomal dominant OMD was the most common phenotype, with one autosomal recessive OMD case identified. Autosomal recessive RP had the latest onset, best visual acuity, and highest refractive error. OMD BCVA declined by ∼0.5 lines/year over a median follow-up of 3.2 years. The researchers commented that the results align “with the clinical observation that patients typically reported their first symptoms in their 30s, with OMD showing the widest age range at onset—from the first to the seventh decade—and atypical RP1L1-retinopathies tending to manifest earlier. Both typical and atypical OMD presented with a marked reduction in visual acuity (VA), followed by a gradual deterioration of approximately 1 line every 2 years, whereas individuals with RP had nearly normal BCVA, which remained stable over the follow-up. Myopia was the most common refractive error, being more pronounced in patients with RP than patients with OMD”.