Researchers at UCL Institute of Ophthalmology, University College London, and Moorfields Eye Hospital, have reported a detailed natural history, retinal imaging dataset and clinical features on CDH23-associated Usher syndrome. Usher syndrome type ID (USH1D) is caused by homozygous or compound heterozygous mutations in the gene encoding cadherin-23 (CDH23), originally mapped to chromosome 10q in 1996. The current study represents the first in-depth retrospective longitudinal study of USH1D patients, indicating that “the natural history of macular involvement is relatively slow”, and the window for intervention may be sufficiently wide to allow different therapeutic strategies expand in due course.
Usher syndrome (USH) is a rare disease, historically categorised into three groups (USH1, USH2, and USH3), dependent on the severity of hearing loss, onset of retinitis pigmentosa (RP) and presence/absence of vestibular dysfunction. (A fourth and atypical subgroup (USH4) are further identified with variants in arylsulfatase G (ARSG) centrosomal proteins (CEP78 and CEP250), and abhydrolase domain-containing protein 12). USH may arise from mutations in at least 9 genes including: MY07A, USH1C, CDH23, PCHD15, USH1G, USH2A, GPR98, WHRN and CLRN. According to the research literature, USH1D accounts for 19% to 35% of USH1 cases and pathogenic variants in CDH23 have reported combined visual and hearing impairment, “making it the third most common cause of dual sensory impairment (DSI) in affected individuals”.
In their current natural history study, 31 patients were identified through a molecularly confirmed USH1D diagnosis from a tertiary referral center (Moorfields Eye Hospital, London, UK) with 40 variants in CDH23 gene (10 of which were being novel). The mean (range, ±SD) age of symptom onset was 10.1 years (range = 1–18, SD = ±4.1) and the most common visual symptoms were nyctalopia (93.5%) and peripheral vision difficulties (61.3%). The mean BCVA at baseline was 0.25 ± 0.22 in the right eyes and 0.35 ± 0.58 LogMAR in the left eyes. The mean annual loss rate in BCVA was 0.018 LogMAR/year over a mean follow-up of 9.5 years. Full-field and pattern ERGs indicated moderate-severe rod-cone or photoreceptor dysfunction and the rate of ellipsoid zone width (EZW) and outer nuclear layer (ONL) thickness loss appeared mild, “suggestive of a wide window of macular preservation”.
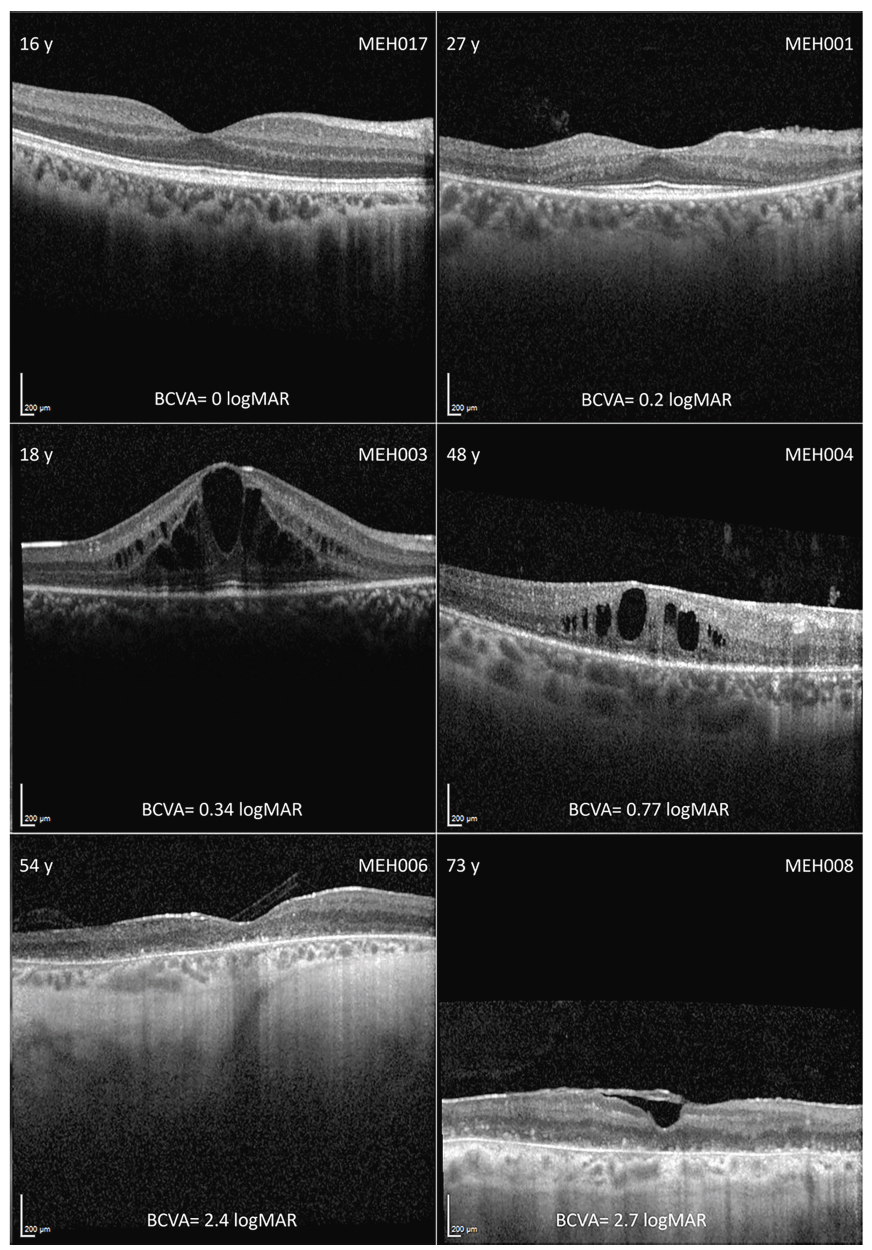
Figure 1: Montage of optical coherence tomography (OCT) imaging in different disease stages. In the left and right top corner are the age (years) and the subject ID, respectively, with the best corrected visual acuity (BCVA) in LogMAR below. Intraretinal cystic spaces were common across the cohort, regardless of disease stage. In late stages, there was diffuse outer retinal and retinal pigmented epithelium atrophy. Subject MEH008 also had an epiretinal membrane. (Open Access – this article is licensed under a Creative Commons Attribution 4.0 International License; de Guimaraes et al. Invest Ophthalmol Vis Sci. 2024;65(8):27. https://doi.org/10.1167/iovs.65.8.27).
Beyond the collection of the valuable data, the researchers commented that, “this is crucial data to improve counselling of affected patients and families and could also be used as a framework for developing novel treatments in the future”. In addition, while active gene therapy studies are in progress worldwide, the UCL team also commented that, “the main restriction for gene therapy is the size of the CDH23 gene, which spans about 10.1 kb and is more than twice the cargo capacity of the current generation of adeno-associated viral vectors, the most commonly used vector for gene therapy. There remains, however, an enormous interest in development of novel vector technology, with some groups developing triple vectors to expand this transfer capacity”.