Researchers at the Department of Ophthalmology and Visual Sciences, University of British Columbia, Canada, have reported the first example of a beta-tubulin 4B gene variant segregating with deaf-blindness. The study, published in Molecular Vision, showed that the novel variant segregating with disease in an extended family establishing autosomal dominant inheritance. The researchers suggested that the phenotype may represent a dominant form of Usher syndrome type 3, characterized by post-lingual sensorineural hearing loss and RP, without vestibular involvement.
Sensorineural hearing and vision loss occur in a heterogeneous group of conditions arising from a range of causes including genetic defects, infections and autoimmune diseases. Genetic causes affect primary cilia, including Usher syndrome (USH), Bardet-Biedl, Waardenburg, Stickler and Alstrom syndromes. Usher syndrome (USH) is a rare disease, historically categorised into three groups (USH1, USH2, and USH3), dependent on the severity of hearing loss, onset of retinitis pigmentosa (RP) and presence/absence of vestibular dysfunction. (A fourth and atypical subgroup (USH4) are further identified with variants in arylsulfatase G (ARSG) centrosomal proteins (CEP78 and CEP250), and abhydrolase domain-containing protein 12). USH may arise from mutations in at least 9 genes including: MY07A, USH1C, CDH23, PCHD15, USH1G, USH2A, GPR98, WHRN and CLRN.
In the current research from the University of British Columbia, four affected members and four unaffected members of a four-generation Canadian family were evaluated showing a phenotype of hearing loss and RP segregated in an autosomal dominant manner:
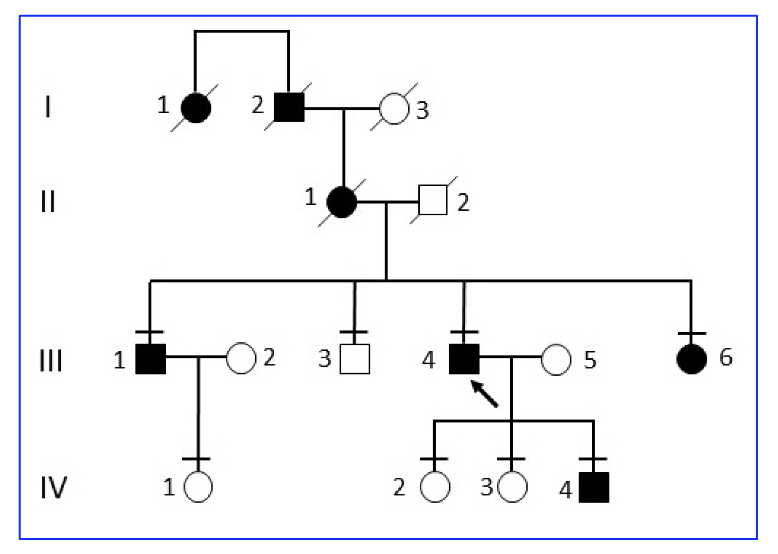
Figure 1. Four-generation family tree segregating autosomal dominant hearing loss and RP. The black symbols represent affected individuals and white symbols represent unaffected individuals. The arrow indicates the proband patient. The line above the symbols denotes family members who were examined. The first two generations were reported by proband as deaf-blind but were not examined or genetically tested. [The research work is licensed under a Creative Commons Attribution-Non Commercial-No Derivatives (http://creativecommons.org/licenses/by-nc-nd/3.0/) cited by Gregory-Evans et al., entitled by: “Mutation of beta-tubulin 4B gene (TUBB4B) causes autosomal dominant retinitis pigmentosa with sensorineural hearing loss in a multigenerational family”, Molecular Vision 2025; 31:175-188 <http://www.molvis.org/molvis/v31/175> ].
Following their clinical testing and analysis, results showed that a panel-based genetic test revealed a heterozygous c.1168C>T, p.Arg390Trp variant in the beta-tubulin 4B gene (TUBB4B), only in affected family members. The researchers commented that, “based on in silico analysis, segregation analysis through the family, and literature evaluation, this variant is likely to be the disease-causing variant inherited in an autosomal dominant manner”, confirmed to be a rare disease variant. To further understand how variants in TUBB4B might cause retinal and hearing loss phenotypes, the researchers used a zebrafish and mouse model and found loss of both rod and cone photoreceptors appeared to be lethal. The study concluded that, “due to the high level of gene conservation, loss of function of Tubb4b/tubb4b leads to early lethality in both mice and zebrafish and suggests that homozygous variants in TUBB4B in humans might also be embryonic lethal. Since the tubb4b zebrafish model demonstrates an eye phenotype unlike mice, this could become the best model to test potential therapeutic options for the retinal phenotype”.