Researchers at the Department of Pathology, University of Oklahoma Health Sciences Centre, USA, have reported a study expanding on the mutation spectrum for inherited retinal diseases (IRDs), including 20 novel variants. The new variants were absent in the literature or not found in large population databases. The results, published by the research team, “clarify and confirm clinical diagnoses, aid in counselling patients on prognosis and family planning, and guide treatment options. This study not only holds promise for affected individuals but also expands the mutation spectrum to guide understanding of IRD.”
According to the researches, IRDs are broadly categorized into three clinical types: photoreceptor disease type, such as retinitis pigmentosa (RP), cone and cone-rod dystrophy (CD/CRD) and Leber congenital amaurosis (LCA); macular disease type, which primarily affects the RPE, such as include Best disease, pattern dystrophy, and Stargardt disease (STGD1), and; finally, the third disease type, with overlapping symptoms, includes IRDs such as choroideremia, optic neuropathy and X-linked retinoschisis (XLRS). In their research, sponsored by the Foundation Fighting Blindness MyRetina Tracker program, the team used buccal samples collected for panel-based sequencing at BluePrint Genetics (BpG), a company based in Helsinki, Finland. The objective was to improve diagnostic accuracy and advance understanding of disease mechanisms, genetic testing, recruiting 103 unrelated patients with an IRD at a single clinical site – the Dean McGee Eye Institute (DMEI) at the University of Oklahoma Health Sciences Center (OUHSC).
A clinical retina specialist performed standard visit assessments, including visual acuity (Snellen chart), slit lamp examination, fundus photography and spectral-domain optical coherence tomography. Following gene panel-based sequencing, variants were classified as pathogenic, likely pathogenic, and variants of uncertain significance (VUS), reported as primary findings on the patients’ genetic report.
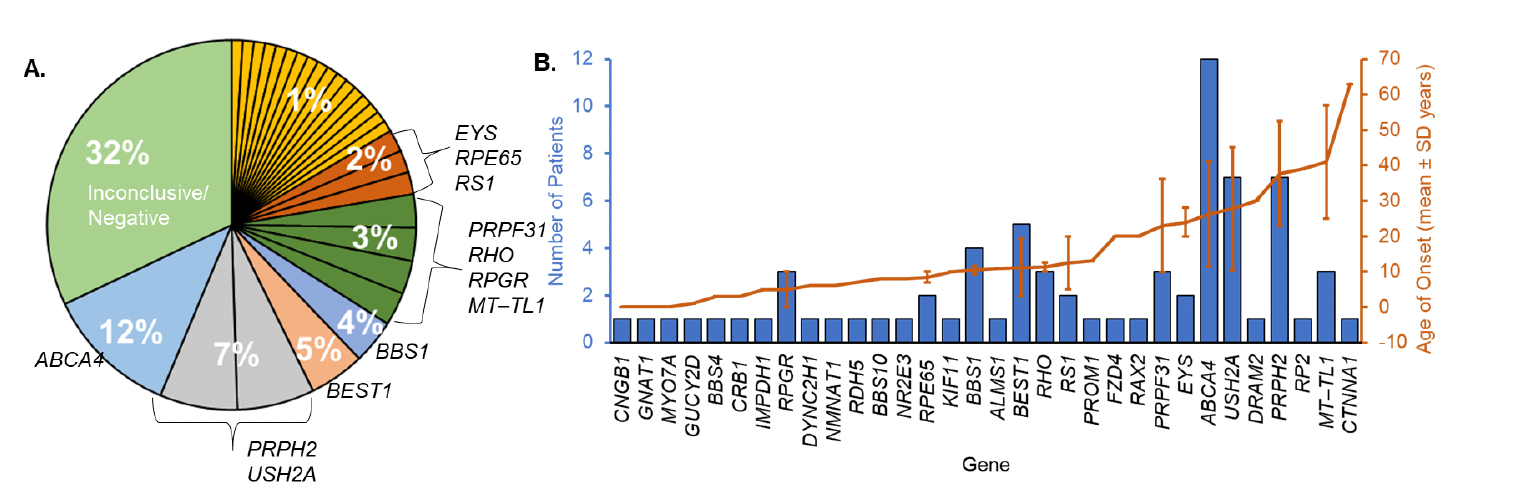
Figure 1: Gene frequency and age of onset. (A) Percentage of cases associated with identified genes. Genes associated with one patient (yellow slices) each were BBS10, BBS4, CNGB1, CRB1, FZD4, GNAT1, IMPDH1, KIF11, MYO7A, NMNAT1, NR2E3, PROM1, RAX2, RDH5, and RP2. (B) Mean ± SD age of onset is indicated by the orange trend line. The number of patients diagnosed per gene is indicated by the blue bars. [Open access article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format when provided to the appropriate credit of: Lynn, J., et al, Expanding the Mutation Spectrum for Inherited Retinal Diseases. Genes 2025, 16, 32. https://doi.org/10.3390/genes16010032].
Following the results, 103 individuals showed causative mutations in 29 genes including the identification of 20 novel variants, six of which were reclassified from variants of uncertain significance (VUS) to likely “pathogenic” by a certified geneticist. ABCA4-related disease was the most common cause of IRD, with the highest variant frequency in their cohort (28%, n = 12 patients). The frequency of PRPH2-related IRD was the second-highest cause of IRD. This study is extremely valuable to clarify inheritance patterns and establish genotype–phenotype correlations concluding that, “our research expands the understanding of the genetic basis of IRD and underscores the clinical and genetic heterogeneity of these conditions, highlighting the need for personalized genetic screening and targeted therapeutic strategies.”