Researchers at Moorfields Eye Hospital, the UCL Institute of Ophthalmology, University College London and the Shiley Eye Institute, University of California, San Diego, has reported two rare nucleotide substitutions at the same genomic location on chromosome 11 (g.61392563 [GRCh38]), upstream of the start codon of the ciliopathy gene “TMEM216”. The ciliopathy gene shows two variants causing a predicted loss-of-function allele with night blindness in the first decade and progressive peripheral field loss. The researchers concluded that, “these variants explain a significant proportion of unsolved cases, specifically in individuals of African ancestry”. Reduced protein expression with TMEM216 potentially leads to abnormal ciliogenesis and photoreceptor degeneration.
A graphical abstract of their publication summarises the outcome from a whole-genome sequence (WGS) data of individuals from a large UK cohort (Genomics England 100,000 Genomes Project) analyzed to uncover the underlying cause of retinal degeneration in previously unresolved cases:
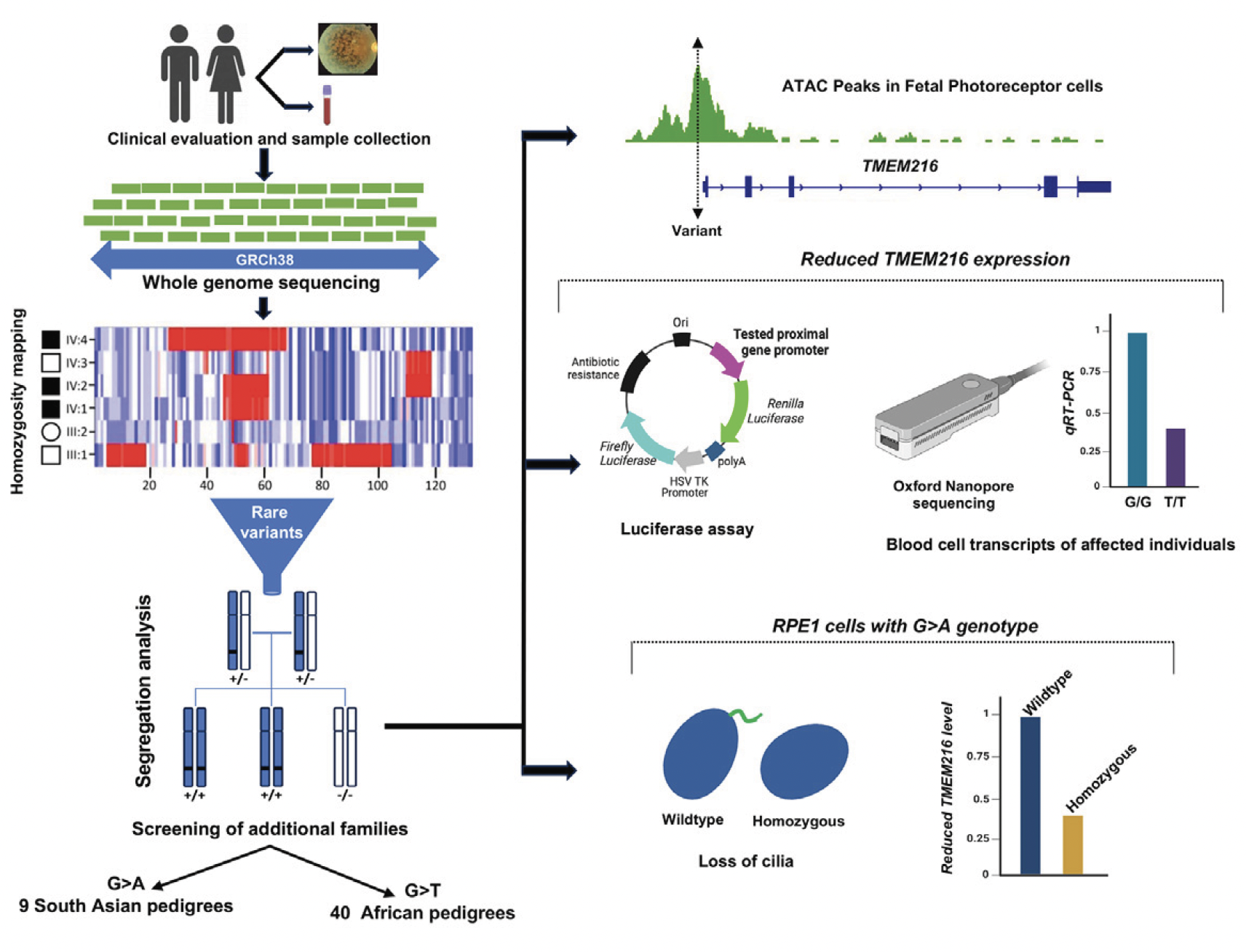
Figure A. Genetic analyses of retinitis pigmentosa (RP)-affected individuals in African and Pakistani populations, along with functional validations, identified two novel pathogenic substitutions in the UTR of TMEM216: c. 69G>T and c. 69G>A. The c. 69G>T variant is estimated to account for about 20% of RP cases in individuals of African ancestry, including African Americans. (Malka et al., Substitution of a single non-coding nucleotide upstream of TMEM216 causes non-syndromic retinitis pigmentosa and is associated, The American Journal of Human Genetics (2024), https://doi.org/10.1016/j.ajhg.2024.07.020; this article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https:// creativecommons.org/licenses/by/ 4.0/).
Published at the American Journal of Human Genetics (Volume 111, Issue 9, p2012-2030, September 05, 2024), their study suggest that there is a lower rate of molecular diagnosis for genetic testing among several ethnicities and this may under-represent available genome datasets. According to their work, unsolved cases in certain patient groups may have missing molecular diagnoses potentially attributed “to non-coding variants, structural rearrangements, variants in unreported RP genes, and incorrect clinical diagnosis”. The analysed variants include a total of 74 individuals affected with RP of African and South Asian ancestry. Variants in TMEM216 and TMEM138 appear to date in Joubert or Meckel syndrome, with a likely loss, or reduced function.
This study showed two non-coding variants in TMEM216 (c. 69 G>A and c. 69G>T), and potentially the underlying cause of non-syndromic RP among 74 affected individuals from 49 families of African (T family) and South Asian (A family) ancestry.
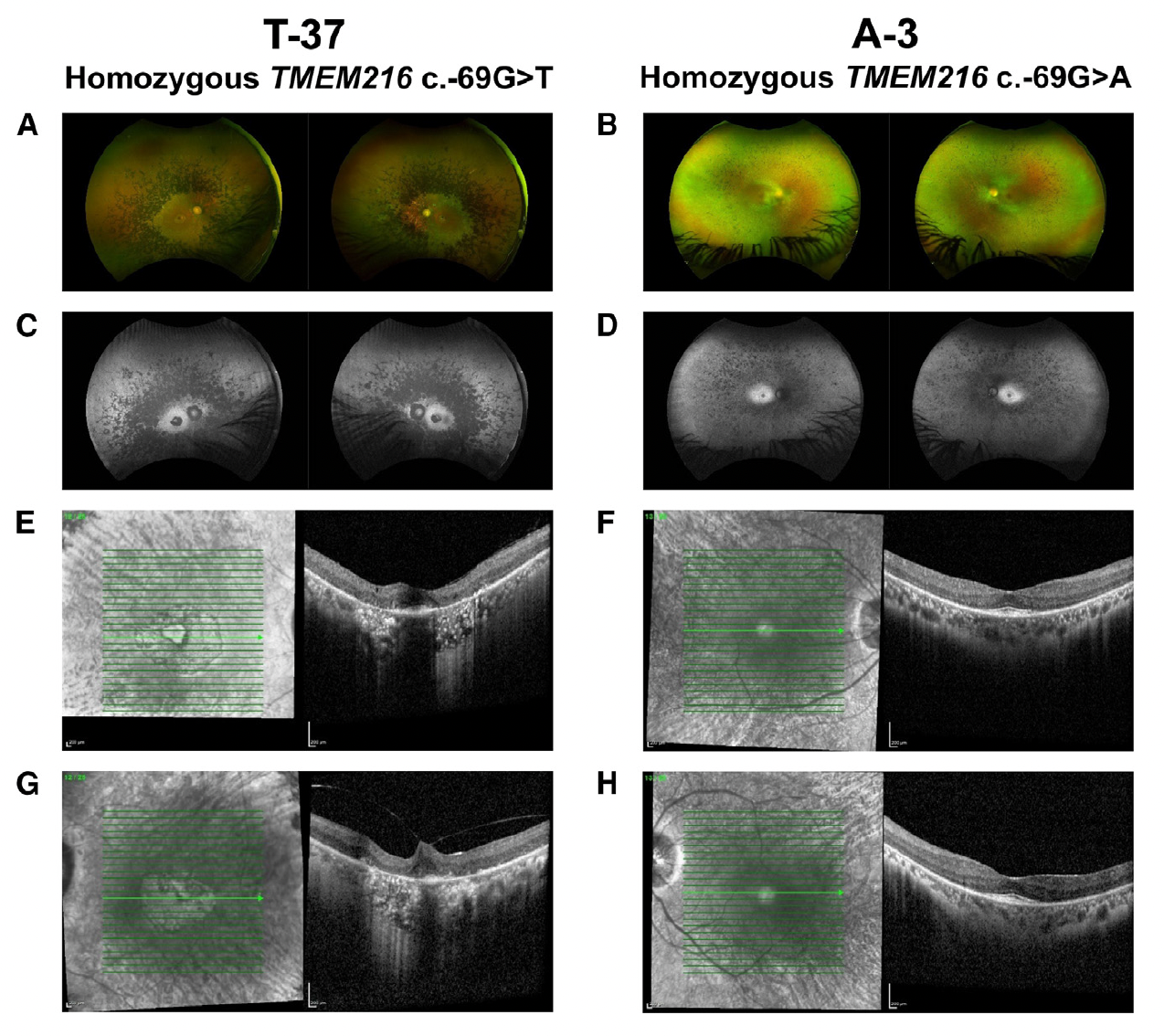
Figure B. Clinical findings for individuals with TMEM216 c. 69G>T and TMEM216 c. 69G>A variants of 72 and 24 years of age, respectively (A–D). En face pseudo color images (A and B) and green (532 nm) autofluorescence (C and D) from an Optos wide-angle fundus camera. The 72-year-old individual shows a greater amount of pigment and further reduction in autofluorescence (A and C). In the 24-year-old individual, there is typical bone-spicule pigment in the peripheral retina. This individual showed loss of autofluorescence, with retention of autofluorescence within a 10-degree area centered on the fovea (B and D). (E–H) En face infrared and OCT images of the right (E and F) and left (G and H) eyes centered on the fovea. The 72-year-old individual showed atrophy of both outer retina and RPE on OCT with some preservation of the foveal layers. In the 24-yearold individual, the region of preserved retinal anatomy on OCT imaging matches the retained autofluorescence observed in (D) (F and H). [Detailed in the open access data available in Malka et al., Substitution of a single non-coding nucleotide upstream of TMEM216 causes non-syndromic
retinitis pigmentosa and is associated, The American Journal of Human Genetics (2024), https://doi.org/10.1016/j.ajhg.2024.07.020; this article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https:// creativecommons.org/licenses/by/ 4.0/).]
Four independent assays showed that the c. 69 G>A or G>T noncoding variants appeared to downregulate the TMEM216 expression, causing reduced activity but not
abrogated by these variants. The researchers propose that “it is likely that the photoreceptor cells are more sensitive to the reduction of gene expression, and therefore the TMEM216 c. 69G>A or G>T variants leading to such gene expression reduction, rather than complete loss of expression, cause specific dysfunction of photoreceptors”. The researchers commented that this phenomenon has been observed in other ciliopathy genes such as CEP290 and the lethal Meckel syndrome (MKS). Finally, the researchers anticipate that such variants “will be a major cause in the remaining genetically unsolved IRD cases” and that that their “findings will open up the possibility of gene-directed therapies such as gene editing or augmentation”.